Purpose: Molecular Dynamics
Latest version: 2.13
License: ![]() Free of use for non-commercial users
Free of use for non-commercial users
Mixed license
Website: http://www.ks.uiuc.edu/
NAMD is a molecular dynamics package.
NAMD implements standard MD capabilities (variety of integrators, thermostats, barostats, force field compatibility, PME electrostatics, time-step integration, replica exchange, free energy calculations, etc.).
NAMD is also available to run on GPU NODES showing up to 8X performance (at same number of cores as std nodes). To perform GPU computations, please contact us.
SLURM Submit script example
More information about the submit script can be found using the Job Script Generator.
#!/bin/bash
#SBATCH -J namd_example
#SBATCH -e namd_example.%j.err
#SBATCH -o namd_example.%j.out
#SBATCH -p std
#SBATCH -n 4
#SBATCH -t 02-00:00
module load apps/namd/2.13
##
# Modify the input and output files!
INPUT_FILE=namd_example.com
OUTPUT_FILE=namd_example.log
cp -r ${SLURM_SUBMIT_DIR}/${INPUT_FILE} ${SCRATCH}
cd ${SCRATCH}
srun `which namd2` ${INPUT_FILE} > ${OUTPUT_FILE}
cp ./${OUTPUT_FILE} ${SLURM_SUBMIT_DIR}
Sbatch options:
The options shown in the example are detailed below. For more information and a more comprehensive list of available options, see the sbatch command page.
- -J: Name for the job's allocation.
- -e: Name of the stderr redirection filename.
- -o: Name of the stdout redirection filename.
- -p: Name of the partition (queue) where the job will be submitted.
-n: Number of tasks.
- -t: Set the job's time limit. If the job don't finish before the time runs out, it will be killed.
Benchmarks
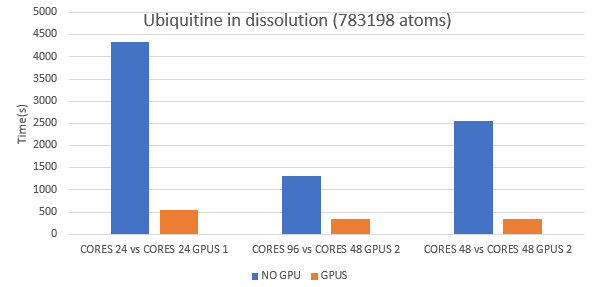
Fig: Comparison between jobs with and without GPGPUs.